 会员登录
会员登录













版权归原作者所有,如有侵权,请联系我们
1806 年,23 岁的德国药剂师 Sertürner 从罂粟中首次分离出单体吗啡,现代天然药物化学研究自此起步。在此基础上,德国化学家 Friedrich Wǒhler 在 1828 年成功实现了尿素的人工合成,这也标志着有机化学学科的正式诞生。可以说,正是人类对生物活性天然产物 (natural product, NPs) 的持续研究促成了有机化学学科的建立。
所谓生物活性天然产物 (NPs) ,其实是自然界长期进化的物质实体,是生物活性物质和实用药物研发的重要源泉。在药物研发进程中,NPs 对于癌症和传染病治疗药物的创新有着巨大贡献。但时至今日,NPs 仍在筛选、分离、表征、优化等各方面存在技术障碍。其中,从复杂混合物中分离 NPs 可谓是最为严峻的挑战之一,这也成为了药物研究的一大瓶颈。
为了解决这一瓶颈, 中南大学湘雅医院药学部刘韶教授团队,创新性地建立了一种可全面挖掘天然药物药效成分的整合分子网络框架 (integrated molecular networking workflow for NP dereplication, IMN4NPD),不仅加快了分子网络中广泛集群的去复制 (dereplication),而且对现有研究方法中经常被忽略的自循环与成对节点提供标注。相关研究成果日前被发表于美国化学会 (ACS) 期刊 Analytical Chemistry。

论文地址:
https://doi.org/10.1021/acs.analchem.3c04746
IMN4NPD:集成多种计算工具,由光谱相似度驱动的分子网络
IMN4NPD 的核心工作原理是由光谱相似度驱动的分子网络。它通过集成并协同 NPClassifier, molDiscovery 和 t-SNE 网络等多种计算工具,从而帮助研究人员快速识别特定类别的化合物,同时还能简化分子网络节点中的标注。
* NPClassifier:一种基于深度神经网络的天然产物结构分类工具
* molDiscovery:一种质谱数据库搜索方法
一般而言,IMN4NPD 的工作流程可分为 3 步:
第一步,对原始 LC-MS 数据进行预处理,以生成分子网络或基于特征的分子网络。随后,基于深度神经网络的 NP 分类工具 SIRIUS,通过 NPClassifier 对复合类进行系统分类。
第二步,该研究通过 GNPS (Global Natural Product Social Molecular Networking),进行了基于 MS/MS 光谱数据库的去复制实验,然后通过 molDiscovery 进行基于计算机数据库的去复制。
第三步,研究人员利用 MS/MS 光谱特征的相似度生成 t-SNE 网络,并对每个节点的化合物进行化学分类,以精确定位、并去复制分布在自循环网络中的特定化合物类别。
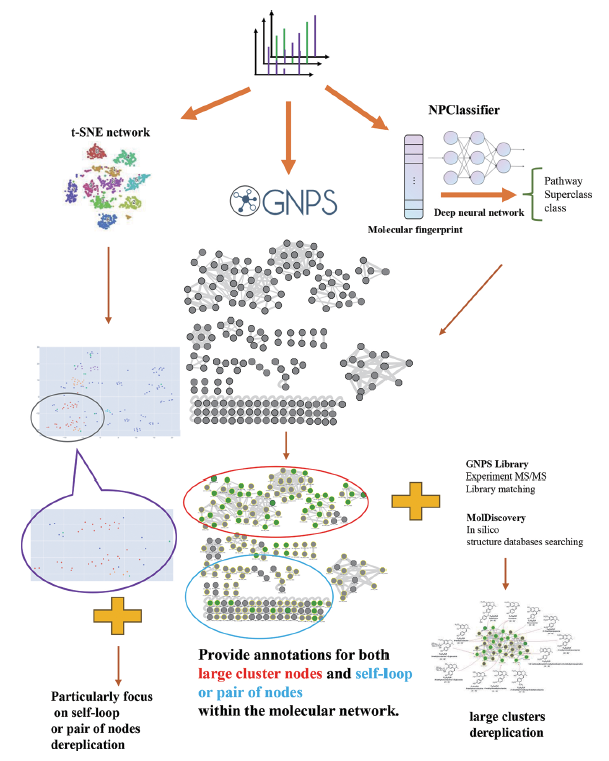
IMN4NPD 工作流程示意图
可用性评估:探索异喹啉类似物,在分子网络中迅速识别特定化合物簇
为了评估 IMN4NPD 工作流程的性能和优势,该研究重新分析了莲子心的乙醇提取物。莲子心是莲蓬中的胚芽部分,是一种富含双苄基异喹啉、单苄基异喹啉和阿朴啡等多种生物碱的中药植物,可用于治疗失眠、遗精、心率失调、高血压等症状。
基于实验性 MS/MS 光谱数据库,该研究最初对分子网络中的单个节点进行化学分类,从而在分子网络中迅速识别出特定的化合物簇,以探索新的异喹啉类似物。在查看了分子网络中每个特征映射的化学分类结果后,研究人员发现,很容易就能找到与异喹啉类似物相对应的某些化合物簇,同时,异喹啉类化合物主要分布在分子网络中的四个簇中。
 异喹啉类化合物分布图
异喹啉类化合物分布图
该研究还发现,通过实验性 MS/MS 光谱数据库(如 GNPS 数据库)只能成功地去复制大型簇中数量有限的特征。因此,该研究采用了最先进的硅学片段算法 molDiscovery 进行结构数据库匹配。这种基于实验和硅学 MS/MS 图谱数据库的去复制方法,增强了在分子网络中、尤其是在大型集群中,及时、方便地标注物质结构的能力。
以单苄基异喹啉生物碱中的集群 A 为例,该集群由 36 个节点组成,其中 MS 数据库只标注了 7 个节点,Structure 数据库标注了 35 个节点,MS 和 Structure 数据库同时标注了 8 个节点。值得注意的是,这其中有一个 m/z 344.1855 节点 (tR=7.6329) 被 MS 结构数据库完全标注,这表明候选结构为 3′-O-methyl-4′-methoxy-N-methylcoclaurine(如上图所示)。
通过进一步分析,该节点连续损失了 NH3CH3、CH3OH 和 H2O,随后发生了环裂解、α 裂解和 β 裂解,分别在 m/z 107.0496、137.0597、151.0757、175.0750、205.1098、235.0752、267.1017、299.1271 和 312.1590 处产生碎片离子。
经 Structure 数据库鉴定,m/z 448.1963(tR = 1.6287)的结点为 N-methylnorcoclaurine 7-O-glucoside。另一个 m/z 312.1593 (tR = 7.3621) 节点则显示了包含 1 个单苄基异喹啉在内的四个候选结构。与 m/z 344.1855 节点 (tR=7.6329) 相比,该节点在 m/z 190.0862 (C11H12NO2) 处存在碎片离子,表明这是一个亚甲基二氧基。
研究结果:基于深度神经网络,从 t-SNE 网络的角度对比三大研究算法
与 MolNetEnhancer 相比,IMN4NPD 采用基于深度神经网络的 NP 分类工具 NPClassifier,来单独分类分子网络中的每个特征,而不是整个簇或分子家族。该研究使用了改进过的余弦相似度计算相似矩阵,并以此生成 t-SNE 网络。同时,该研究还通过 NPClassifier 基于每个节点的 MS/MS 光谱数据对其进行分类,并将这些分类映射到 t-SNE 网络中。
在传统的分子网络观点中,异喹啉一般由三个大簇 (簇 A-C) 和一个小簇 (簇 D) 共同组成。从 t-SNE 网络的角度来看,很明显,异喹啉的四个集群节点被紧密分组,形成了不同的集群区域。但值得注意的是,从 t-SNE 网络的角度来看,分子网络中的簇 A 可以进一步分为两个更小的簇。此外,t-SNE 可以有效定位异喹啉类节点,从而大大减轻了相关节点的结构解析工作。
 异喹啉在 t-SNE 图谱中的四个聚类区域
异喹啉在 t-SNE 图谱中的四个聚类区域
修正余弦相似度方法面对多种学修饰的化合物光谱存在局限性,该研究还选择了 Spec2Vec 和 MS2DeepScore 等相似度算法,并以此生成 t-SNE 网络。基于 Spec2Vec,异喹啉依然在分子网络中形成四大簇区。
但基于 MS2DeepScore,异喹啉的大簇 A 和 B 的节点间隔很近,形成了几个聚类区域,但大簇 C 中的节点分散在了整张图中,这为后续分析带来了挑战。
 多种光谱相似度算法生成的 t-SNE 图谱比较
多种光谱相似度算法生成的 t-SNE 图谱比较
一个有趣的现象是,m/z 296.1646节点 (tR = 11.54) 在修正余弦相似度和 MS2DeepScore 相似度的 t-SNE 图中,均远离异喹啉相关的节点聚类区域,但在基于 Spec2Vec 光谱相似度的 t-SNE 图中,该节点与大簇 A 的聚类区域相邻。这类自换节点可能代表了一类异喹啉化合物,在进一步比较后可确认该节点是阿朴啡类生物碱。
因此,化合物化学分类和 t-SNE 网络,可分别提供关于特征的不同信息,一定程度上减少了假阴性的出现。
此外,基于 Spec2Vec 光谱相似度的 t-SNE 网络,大簇 A 附近存在 m/z 298.1438 (tR = 7.02) 和 m/z 298.1438 (tR = 7.60) 两个节点,这两个节点是分子网络中的自换节点和对节点。尽管没有被归类为异喹啉化合物,但它们与异喹啉大簇 A 结构相似。进一步分析可知,m/z 298.1438 (tR = 7.02) 是一种已知的阿朴啡类生物碱——nornuciferidine,m/z 298.1438 (tR = 7.60) 也显示出与 nuciferine 和 nornuciferidine 相类似的阿朴啡类生物碱。
通过对以上三个节点的研究发现,它们都属于阿朴啡类生物碱,这与单苄基异喹啉类生物碱不同。在利用修正余弦相似度和 MS2DeepScore 相似度时,这三个节点远离单苄基异喹啉类生物碱相关节点的聚类区域大簇 A,但基于 Spec2Vec,这三个节点却可在大簇 A 附近被发现。
这种差异表明 Spec2Vec 光谱相似性在准确捕捉异喹啉类化合物相似结构方面的卓越能力。
人工智能在天然产物研究中的应用加速
近年来,受益于各种现代技术的迅猛发展,在天然生物活性分子的研究中涌现出了一大批基于 LC-MS/MS 和 NMR 技术,并集成生物信息学、代谢组学、计算机科学等多学科技术手段的新策略和新方法。尤其是,随着人工智能和机器学习算法开始融入天然产物研究工作,进一步为研究人员带来了新一轮的生产力革命。
最初,人工智能的应用集中在有机分子的数字化,以及使用降维技术绘制 NP 化学空间图。后来,研究者通过开发机器学习二元分类器来预测 NP 的生物功能。如今,神经网络架构开始被用于基因组挖掘和分子设计,深度学习算法在药物发现和分子信息学领域越来越受欢迎。
所以,我们可以看到,产学研各界近年来均加快了相关研究的步伐。2022 年,国家超级计算广州中心就联合中山大学、星药科技、美国麻省理工学院和佐治亚理工学院,基于「天河二号」的强大计算和存储能力,提出了一种深度学习驱动的生物逆合成路径导航工具 BioNavi-NP。
而在企业界,天然产物的研究也在不断加速。2023 年,天士力医药集团与华为云达成合作,双方将结合天然产物现代化研究数据等,共建中医药领域垂直大模型。
然而,天然产物数据库仍然是科研进程中的一大挑战。当前,全世界主流的天然产物数据存储库,包括生物合成基因簇的最小信息 (MIBiG)、天然产物图谱 (NP 图谱)、全球天然产物分子网络 (GNPS)、天然产品磁共振数据库 (NP-MRD) 等,但这些数据库的覆盖率较低,并且存在较为常见的数据错误问题,这些都阻碍了人工智能在天然产物药物发现方面的进展。
近年来,中国科学家屠呦呦、日本科学家大村智和爱尔兰科学家 William C. Campbell 等多位研究者因在天然产物全合成方面的成就获得了诺贝尔化学奖提名。毫无疑问,随着天然产物的重要性不断凸显,人工智能在天然产物研究方面的融合也即将按下加速键。
欢迎扫码关注深i科普!
我们将定期推出
公益、免费、优惠的科普活动和科普好物!
















- 参加最新科普活动
- 认识科普小朋友
- 成为科学小记者




